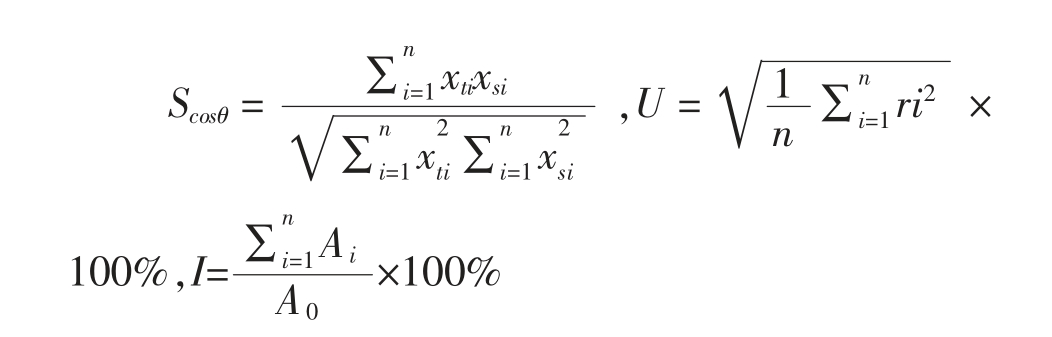毛细管区带电泳在牛乳蛋白掺假检测中的应用及方法优化
方 冰1,2 张 昊2 郭慧媛2 任发政2*
(1 中国农业大学 北京食品营养与人类健康高精尖创新中心 北京1000832 中国农业大学食品科学与营养工程学院 教育部北京市共建功能乳品重点实验室 北京100083)
摘要 目的:利用毛细管区带电泳技术,进行牛乳掺假检测,解决原料乳收购环节,快速判定是否有非乳源蛋白的掺杂的难题。方法:本文通过优化毛细管区带电泳分离条件中的缓冲体系及其pH、进样条件、毛细管长度、分离电压,最终建立适于牛乳蛋白的最佳检测条件。结果:适于牛乳蛋白的最佳检测条件为60 cm 毛细管柱,pH 2.75 的磷酸盐缓冲液,6.895 kPa、10 s 的压力进样方式及25 kV 的分离电压。利用优化的方法建立牛乳蛋白中14 种毛细管区带电泳图谱,以及常见的掺假蛋白——大豆分离蛋白和水解胶原蛋白的检测方法,明确其在原料乳中的特征定性峰、定量回归方程(相关性指数均大于0.996)及其最低检测限(4.5%和4.8%)。结论:本文通过优化牛乳蛋白的毛细管区带电泳分离条件,最终建立起牛乳蛋白的毛细管区带电泳图谱,并将其应用于原料乳蛋白组成和原料乳蛋白掺杂测定,为原料乳收购环节的乳蛋白掺假检测提供一种快速、可靠的方法。
关键词 毛细管区带电泳;大豆分离蛋白;水解胶原蛋白;掺杂检测
在食品生产过程中加入相对廉价的原料是最常见的掺杂现象,这同样存在于乳制品的生产过程中。一些价格相对低廉的动、植物蛋白常被添加到乳中作为乳蛋白替代物,这一现象在原料乳收购环节尤为明显。鉴于此,乳品加工企业在原料乳收购环节进行牛乳蛋白的掺假检测十分重要。
目前牛乳蛋白掺假检测的方法主要有聚丙烯酰胺凝胶电泳、高效液相色谱法、酶联免疫吸附法和毛细管电泳4 种。聚丙烯酰胺凝胶电泳主要是根据待测蛋白质的分子质量和等电点的差异实现分离、鉴定,通常结合两种鉴别原理,即利用二维电泳进行水牛乳[1]、山羊乳和绵羊乳中蛋白的鉴别[2-3]。然而,蛋白质分子在加工过程中容易发生水解以及糖基化、磷酸化等修饰变化,引起分子质量及等电点的变化;此外,蛋白质分子在电泳条带中的分布,极易受到凝胶的浓度、缓冲液体系、凝胶迁移速度等条件的影响,而分子质量的确证往往依赖于标准蛋白条带的参照,使得该法主观性较大;加上一些低峰度的蛋白质较难检出的限制,聚丙烯酰胺凝胶电泳法较难应用。高效液相色谱法常用于乳蛋白组成的分离鉴定[4-8],在掺假检测方面亦有报道,如鉴别脱脂奶粉中掺加的植物蛋白(大豆蛋白和豌豆蛋白)[9];用β-Lg 作为标志物,检测水牛奶及其干酪中掺加的牛奶[10]。然而,蛋白质具有亚基结构,立体构型及构象复杂,有可能发生有机流动相中蛋白质的变性或被测组分的共洗脱现象,降低分离效率及鉴别能力[11]。此方法所需的色谱柱和色谱纯有机溶剂,导致检测方法的高成本进一步降低了其在掺假中的推广应用。酶联免疫吸附法具有高特异性和高灵敏度,特别适用于乳中低含量蛋白的分析[11],如利用牛乳β-酪蛋白的特异性抗体进行山羊乳及其制品中牛乳掺假的鉴定[12]。该法常受限于掺假蛋白特异性抗体的缺乏,以及加工过程中的热处理带来的目标蛋白抗原表位的改变。相较于前面3 种方法,毛细管电泳因没有固定相,故避免了固定相或有机改性剂带来的蛋白构象变化[13];而且毛细管电泳只需很少量的样品和缓冲溶液,显著降低了溶剂的消耗[14]。毛细管电泳技术因高分辨率、 高分离度和快速简便的优点,故在乳蛋白研究领域,尤其是牛乳蛋白的掺假鉴定中显示出极大的应用前景。
毛细管电泳技术首先解决了乳中低丰度蛋白的分离鉴定的问题。α-乳白蛋白在牛乳中的含量较低(占总蛋白的3.7%~5.0%),加工过程还会导致进一步的降解,较难检测。Gutierrez 等[15]利用毛细管电泳建立一种分离和鉴定奶粉中α-乳白蛋白的方法,检测限低至0.01 mg/mL。其次,毛细管电泳解决了同一种蛋白不同亚型的分离鉴定的难题。β-乳球蛋白存在多达8 种的基因变异体。Paterson 等[16] 基于特定pH 下3 种变体的电荷差异,通过优化缓冲溶液的种类、浓度、pH 等参数,实现了β-乳球蛋白A、B、C 变体的分离和鉴定。第三,毛细管电泳可以实现在贮藏、加工过程中发生水解等变化后的乳蛋白分离鉴定。可分析原料乳和UHT 乳在贮藏过程中由蛋白水解作用带来的乳清蛋白占总蛋白的比例变化[17-19]。Recio 等[20]采用毛细管区带电泳方法,分别用血纤维蛋白溶酶和凝乳酶处理牛乳和干酪中的酪蛋白,跟踪其水解过程,鉴别了主要的酪蛋白水解产物,同时分离出凝乳酶作用于αs1-酪蛋白产生的αs1-Ⅰ-酪蛋白和αs1-酪蛋白 f(1-23)以及凝乳酶作用于κ-酪蛋白产生的para-κ-酪蛋白和酪蛋白巨肽。文献[21]分析干酪和乳清蛋白产品中的蛋白质及其降解产物。
鉴于以上优点,毛细管电泳在乳制品掺假检测方面显示出极大的应用前景[8,22-27]。牛乳价格低廉,常被加入山羊乳和绵羊乳中。Recio 等[23]利用毛细管电泳检测绵羊乳制成的干酪中掺加的山羊乳或牛乳,山羊乳和牛乳的掺入量检测限分别为2%和5%。文献[25]基于αs1-酪蛋白的电泳峰,用毛细管区带电泳检测山羊乳中添加的牛乳,检测限低至1%。本文利用毛细管区带电泳方法,通过优化分离缓冲体系及其pH、进样条件、毛细管长度、电压等条件,利用优化的方法建立原料牛乳的蛋白电泳图谱,确定电泳图谱中出现的牛乳蛋白共有峰,鉴定每个电泳峰代表的乳蛋白种类,并用相似度和含量相似度对获得的电泳图谱进行评价。
1 材料与方法
1.1 试验材料
原料牛乳及巴氏杀菌牛乳采自北京三元集团绿荷渠头牧场,α-乳白蛋白 (α-La)、β-乳球蛋白(β-Lg)、α-酪蛋白 (α-CN)、β-酪蛋白 (β-CN)和κ-酪蛋白(κ-CN)标准品及二硫苏糖醇购于美国Sigma 公司,电泳用尿素购于美国Amresco 公司,大豆分离蛋白(大于90%)购于河南正兴食品添加剂有限公司,水解胶原蛋白(大于95%)购于郑州蓝天生物科技有限公司。
1.2 仪器设备
P/ACE MDQ 毛细管电泳仪及未涂层毛细管柱,美国Beckman 公司;低温离心机,美国Thermo Scientific 公司;超纯水仪,ELGA Lab Water 公司;KH5200DB 型超声波清洗器,昆山禾创超声仪器有限公司;精密酸度计,北京哈纳科仪科技有限公司。
1.3 方法
1.3.1 牛乳样品的采集及制备 试验所用牛乳样品为超市购买的巴氏杀菌牛乳和从奶牛场采集的原料牛乳。原料牛乳在同一天采集自同一奶牛场的32 头荷斯坦乳牛,将采集得到的原料牛乳进行体细胞数和蛋白含量测定,从中选取16 份原料牛乳作为被测样品(体细胞数<25 万个/mL,蛋白含量为2.90~3.20 g/100 mL)。将待测牛乳样品在4℃条件下3 100 g 离心20 min 除去脂肪,取一定体积的下层脱脂乳,与样品缓冲液按1∶4(体积比)比例混合,室温放置1 h 后用0.45 μm 的水系滤膜过滤,备用。
1.3.2 毛细管区带电泳分离条件的优化 采用非涂层毛细管(内径50 μm)进行试验,设置柱温为35 ℃,紫外检测波长为214 nm,选择样品缓冲液pH 为8.0,羟丙基甲基纤维素为添加物[28]。优化参数包括进样压力和时间(0.5,1.0 psi,5,10s),毛细管柱长度(50,60 cm),电泳缓冲液体系(柠檬酸盐缓冲液、磷酸盐缓冲液)和缓冲液pH(2.5,2.75,3.0),分离电压(15,20,25 kV)。其中,所用缓冲液的配制方法如下:
1)柠檬酸电泳缓冲溶液 0.05%羟丙基甲基纤维素,6 mol/L 尿素,20 mmol/L 柠檬酸三钠,用32 mmol/L 柠檬酸溶液调节pH 至3.0,2.75 和2.5,定容。用0.45 μm 水系滤膜对配制好的溶液进行过滤,超声,备用。
2)柠檬酸上品缓冲液 40 mmol/L 柠檬酸三钠,0.1%DTT,6 mol/L 尿素,用NaOH 溶液调节pH至8.0,定容。用0.45 μm 水系滤膜对配制好的溶液进行过滤,超声,备用。
3)磷酸盐电泳缓冲溶液 20 mmol/L 磷酸二氢钠,0.05%羟丙基甲基纤维素,6 mol/L 尿素,用磷酸溶液调节pH 至3.0,2.75 和2.5,定容。用0.45 μm 水系滤膜对配制好的溶液进行过滤,超声,备用。
4)磷酸盐上样缓冲液 40 mmol/L 磷酸二氢钠,0.1%DTT,6 mol/L 尿素,用NaOH 溶液调节pH至8.0,定容。用0.45 μm 水系滤膜对配制好的溶液进行过滤,超声,备用。
1.3.3 牛乳样品及标准品的毛细管电泳测定 取处理好的牛乳样品1 mL 于进样瓶中,按照优化好的电泳条件进行测定,每个样品重复测定两次。毛细管使用前的活化以及实验过程中毛细管的清洗程序如下:
1)毛细管使用前的活化程序为:调节压力至20 psi,先 后 用0.1 mol/L NaOH 溶 液、0.1 mol/L HCI 溶液和超纯水冲洗10,5 和2 min;
2)样品测定开始前,毛细管的冲洗程序为:调节压力至70 psi,先后用超纯水、0.1 mol/L NaOH 溶液、超纯水、电泳缓冲液分别冲洗1,2,2和5 min;
3)样品测定结束后,毛细管的冲洗程序为:调节压力至70 psi,用0.1 mol/L NaOH 溶液冲洗10 min,再调节压力至50 psi,先后用0.1 mol/L HCI 溶液和超纯水冲洗10 min 和2 min。
1.3.4 毛细管区带电泳的方法学评价
1)重复性试验 将同一牛乳样品连续进样6 次,获得毛细管区带电泳图谱,计算图谱中各蛋白峰相对迁移时间和峰面积的相对标准偏差(RSD),考察方法的精密度。
2)回收率试验 将牛乳蛋白标准品加入到牛乳样品中,获得毛细管区带电泳图谱,计算加标回收率,考察方法的准确度。
1.3.5 牛乳蛋白毛细管区带电泳图谱的建立 将试验样品按照优化好的电泳条件进行测定,得到各自的电泳图谱,并按以下步骤对电泳图谱进行处理。对获得的一系列电泳图谱进行评价并得到牛乳蛋白毛细管区带电泳对照图谱。选择各谱图中峰共有率不低于70%的作为确定指纹峰的依据。
采用牛乳蛋白标准品对电泳图谱中的主要乳清蛋白 (α-乳白蛋白和β-乳球蛋白)和酪蛋白(α-、β-和κ-酪蛋白)进行标定。并结合已有的文献报道,确定出这几种蛋白峰之外的电泳峰所代表的乳蛋白种类[29]。
1.3.6 掺杂牛乳样品的制备 外源蛋白溶液的准备:各自称取0.03 g 大豆分离蛋白和水解胶原蛋白,分别溶于1 mL 超纯水中,充分搅拌使溶解平衡(注:鉴于蛋白本身的纯度,大豆分离蛋白和水解胶原蛋白分别按照蛋白含量为90%,95%和85%进行计算)。将所得的外源蛋白溶液与样品缓冲液按1∶4(V/V)比例混合,室温下放置1 h,0.45 μm 滤膜过滤,备用。在脱脂牛乳中分别添加50%,40%,30%,20%,10%,5%和2%(体积比)的大豆分离蛋白溶液和水解胶原蛋白溶液,得到掺杂的牛乳样品,再将掺杂样品与样品缓冲液按1∶4(V/V)比例混合,室温下放置1 h,0.45 μm 滤膜过滤,备用。
2 结果与讨论
2.1 毛细管区带电泳分离条件的优化
2.1.1 进样条件的选择 在一定时间范围内,进样时间越长,检测的灵敏度越高;但当进样时间超过10 s 时,出现电泳峰展宽,峰拖尾等峰形变差的负面效应,还有部分峰重叠,分离度下降[30]。因此选择进样条件为压力:0.5,1.0 psi 和时间:5,10 s,进行正交试验。试验结果表明,在一定范围内,同时增加进样压力和进样时间,使电泳图谱中最高峰的纵坐标响应值从25 mAU 增加到了55 mAU,得到显著提高。因此选择进样条件为1.0 psi,10 s。
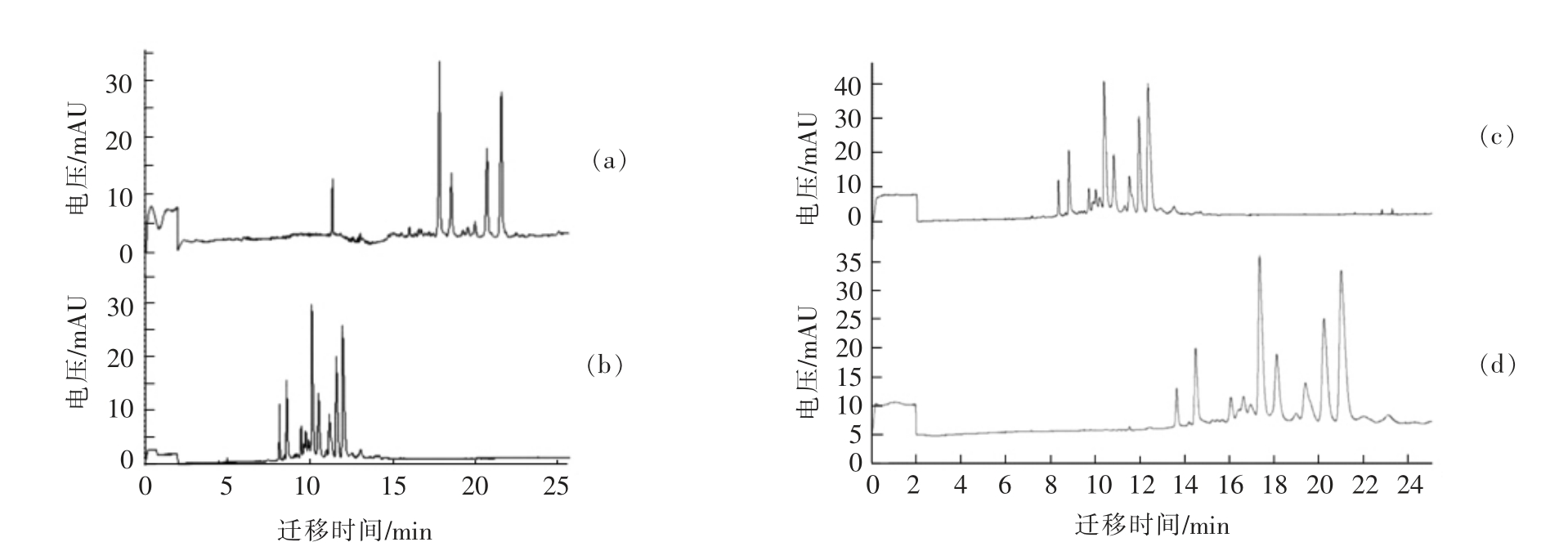
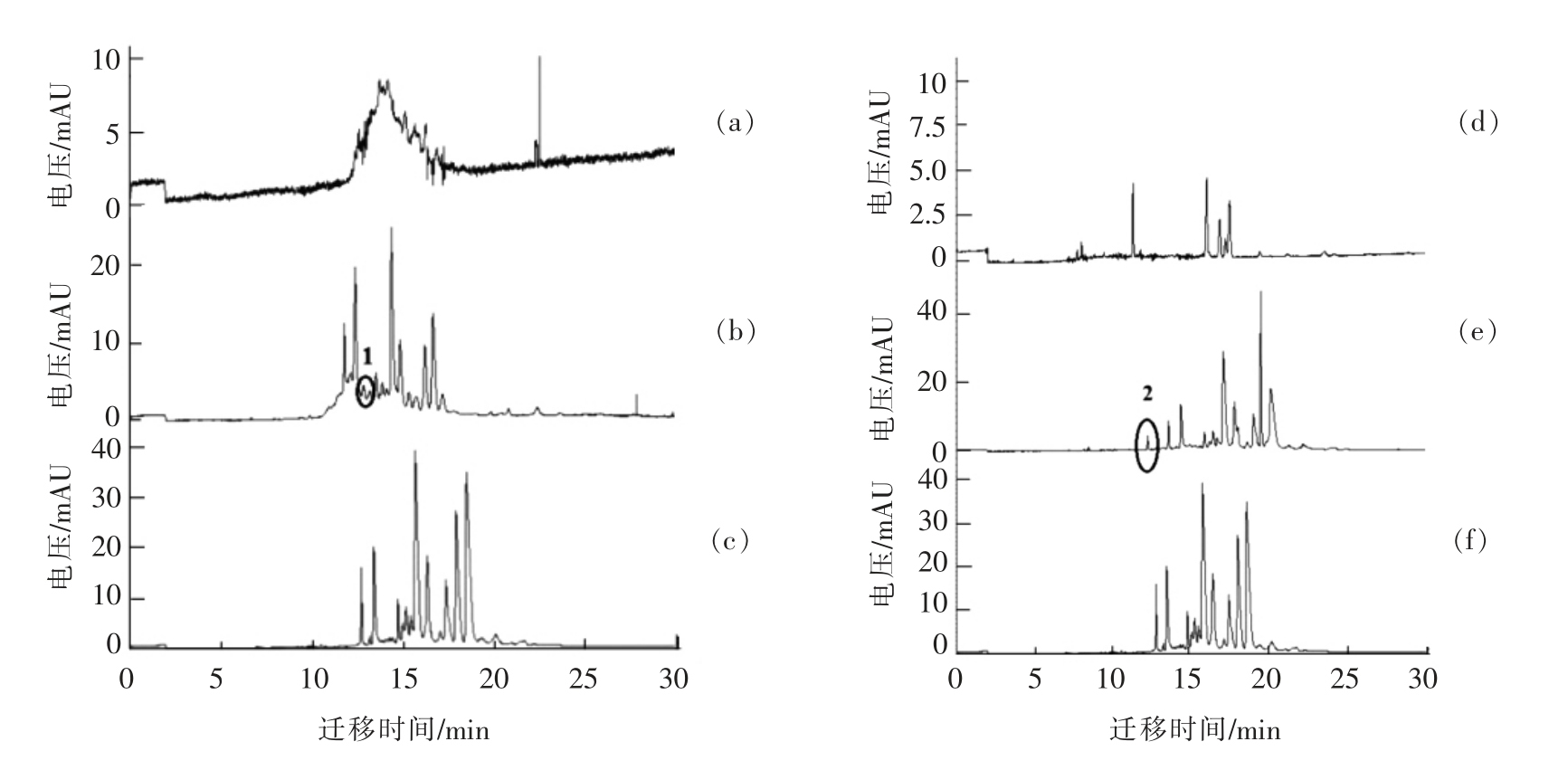
2.1.2 缓冲体系的选择 如图1所示,采用柠檬酸盐缓冲体系(A)作为缓冲溶液时,得到的样品电泳图谱出峰不全,低含量蛋白的电泳峰响应值较低,并且有图谱基线不稳的情况出现;而用磷酸盐缓冲体系(B)作为电泳缓冲液和样品缓冲液,得到的样品电泳图谱分离效果较好。因此本试验选用磷酸盐缓冲体系。
2.1.3 毛细管长度的选择 毛细管长度的选择取决于所检测体系的复杂程度以及对于分离度的要求。复杂体系一般选用较长的毛细管,毛细管长度越长,分离度越好,但随着毛细管长度的增加,样品的迁移时间也随之变长,导致检测所需时间延长。巴氏杀菌牛乳在不同长度的毛细管下的电泳图如图1所示,对于本试验中的牛乳样品,长度为60 cm(D)的毛细管相对于50 cm(C)的毛细管分离效果更好,因此选择长度为60 cm 的毛细管进行试验。
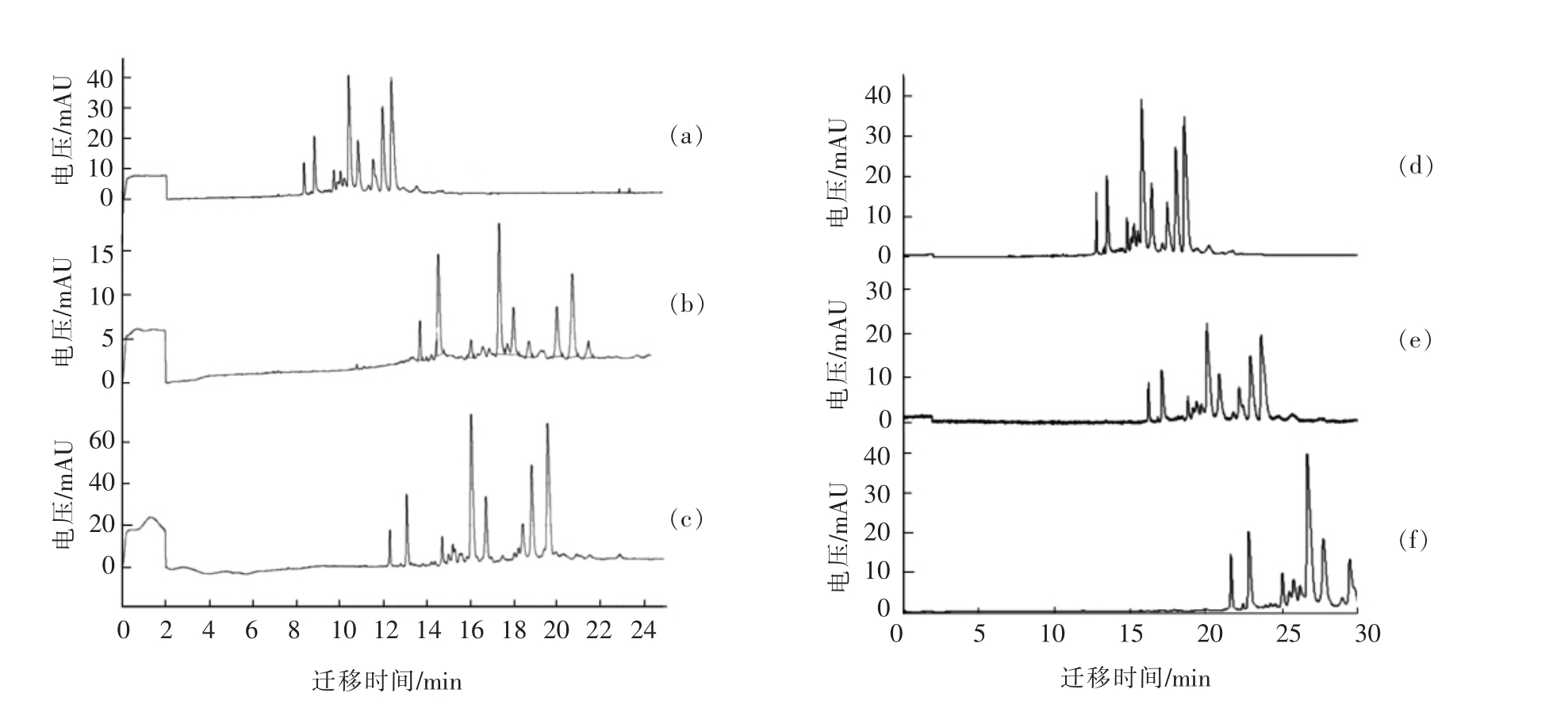
2.1.4 电泳缓冲液pH 的选择 巴氏杀菌牛乳在不同pH 条件下的毛细管电泳图如图2所示。试验发现,随着电泳缓冲液pH 的降低,牛乳蛋白的分离度增加,但是pH 的降低会带来更高的离子强度,从而使电渗流增加,焦耳热增多,导致灵敏度下降。并且当电泳缓冲液的pH 降为2.5 时(图2c),运行电流显著增大,且电流不稳定。因此,选择电泳缓冲液的最佳pH 为2.75(图2b)。
2.1.5 分离电压的选择 巴氏杀菌牛乳在不同的分离电压下的毛细管电泳图如图2所示。当分离电压降低时,电流随之下降,减小了焦耳热的产生。但是随着分离电压的降低,样品组分的出峰时间明显延后。当分离电压分别设置为25 kV (图2d)、20 kV(图2e)和15 kV(图2f)时,被测样品的电泳图谱中首个样品峰的出峰时间分别为12.733,16.204 min 和大于20 min,且当分离电压为15 kV 时,样品的总分析时间大于30 min。因此选择最佳分离电压为25 kV。
综合以上结果,本文中最终确定的毛细管电泳的最佳检测条件为:未涂层毛细管柱(60 cm×50 μm I.D.,有效长度:57 cm),磷酸盐缓冲体系,电泳缓冲液pH 2.75,样品缓冲液pH 8.0,分离电压25 kV,压力进样:1.0 psi,10 s,柱温35 ℃,紫外检测波长214 nm。
2.2 牛乳蛋白标准曲线及方法评价
将经处理的各标准溶液按所选条件测定,并记录数据,以浓度为横坐标,峰面积为纵坐标绘制标准曲线。以各蛋白的峰面积A 对浓度c 进行线性回归,得到各蛋白的线性回归方程、线性范围及相关系数(表1)。5 种主要牛乳蛋白的相对迁移时间及峰面积的相对标准偏差均小于5%,加标回收率均在85%~105%之间,表明方法的精密度和准确度较好。
2.3 牛乳蛋白毛细管区带电泳图谱的建立
2.3.1 电泳图谱指纹峰的确定 对收集的原料乳样品进行分析,记录各自的电泳图。通过计算样品图谱中各组分峰的共有率,得到共有峰14 个,各样品共有峰的迁移时间如表2,各共有峰迁移时间的相对标准偏差均在5%以内。
表1 各蛋白标准品的回归方程、相关系数、线性范围及对应的重复性和回收率
Table 1 The regression equation,correlation coefficient,linear range and the corresponding repeatability and recovery of each protein standards
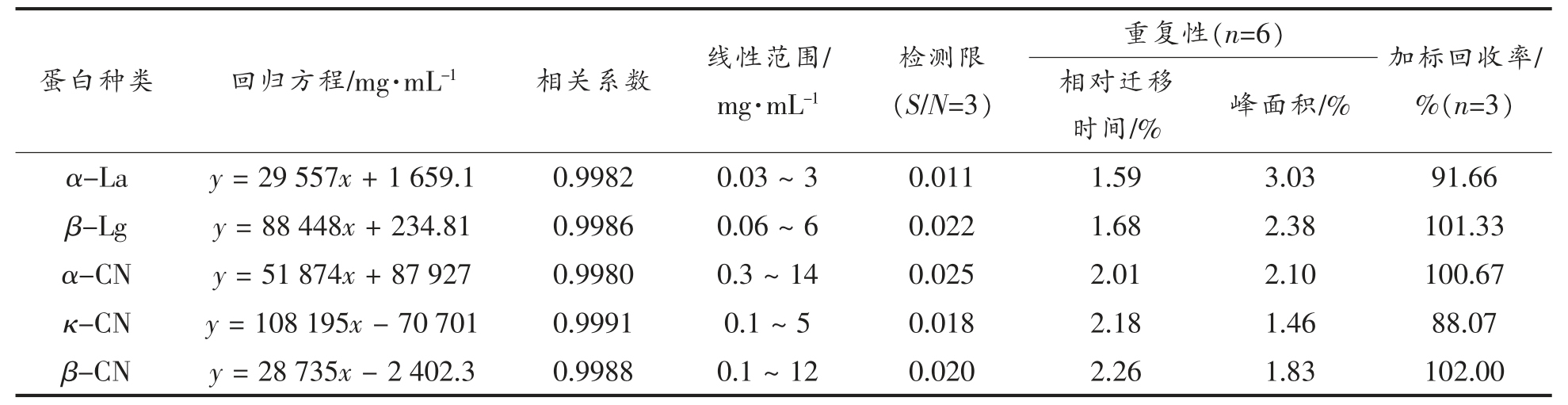
蛋白种类 回归方程/mg·mL-1 相关系数 线性范围/mg·mL-1检测限(S/N=3)重复性(n=6) 加标回收率/%(n=3)相对迁移时间/% 峰面积/%α-La y = 29 557x+1 659.1 0.9982 0.03 ~ 3 0.011 1.59 3.03 91.66 β-Lg y = 88 448x+234.81 0.9986 0.06 ~ 6 0.022 1.68 2.38 101.33 α-CN y = 51 874x+87 927 0.9980 0.3 ~ 14 0.025 2.01 2.10 100.67 κ-CN y = 108 195x - 70 701 0.9991 0.1 ~ 5 0.018 2.18 1.46 88.07 β-CN y = 28 735x - 2 402.3 0.9988 0.1 ~ 12 0.020 2.26 1.83 102.00
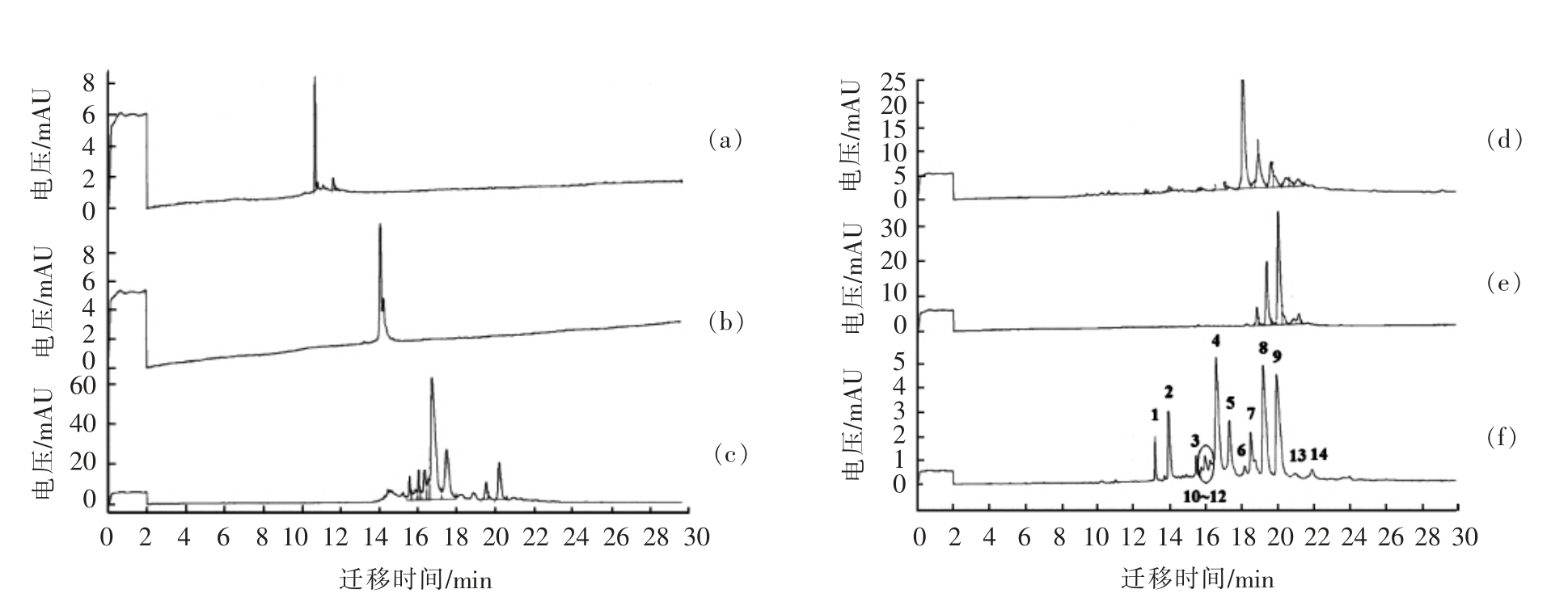
2.3.2 电泳图谱指纹峰的标定 采用牛乳蛋白标准品对电泳图谱中的主要乳清蛋白 (图3a,α-La和图3b,β-Lg)和酪蛋白(图3c-e,分别为α-CN、β-CN 和κ-CN),结合它们各自的出峰顺序,对原料乳电泳图谱进行定性,如图3所示。部分蛋白(图3f 中峰10~14)峰因缺乏相应标准品,根据已有报道,确定电泳图谱中其余电泳峰所代表的乳蛋白种类(图3f)。
表2 牛乳蛋白共有峰的迁移时间
Table 2 The migration time of the peaks in common
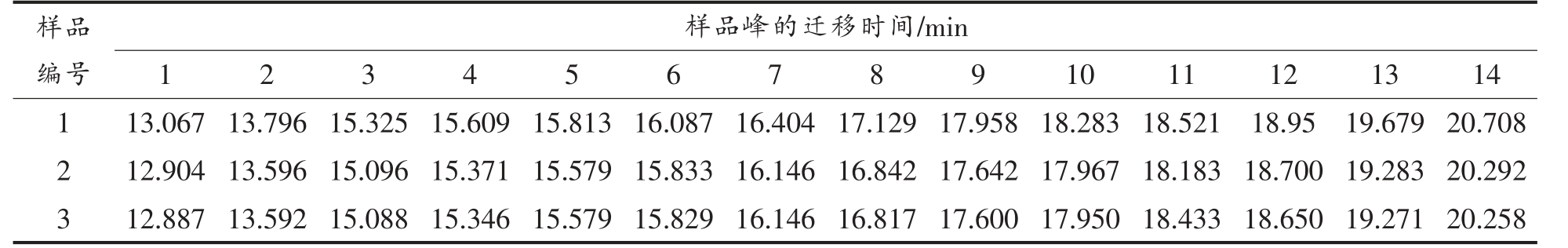
样品编号样品峰的迁移时间/min 1 2 3 4 5 6 7 8 9 10 11 12 13 14 1 13.067 13.796 15.325 15.609 15.813 16.087 16.404 17.129 17.958 18.283 18.521 18.95 19.679 20.708 2 12.904 13.596 15.096 15.371 15.579 15.833 16.146 16.842 17.642 17.967 18.183 18.700 19.283 20.292 3 12.887 13.592 15.088 15.346 15.579 15.829 16.146 16.817 17.600 17.950 18.433 18.650 19.271 20.258
(续表2)
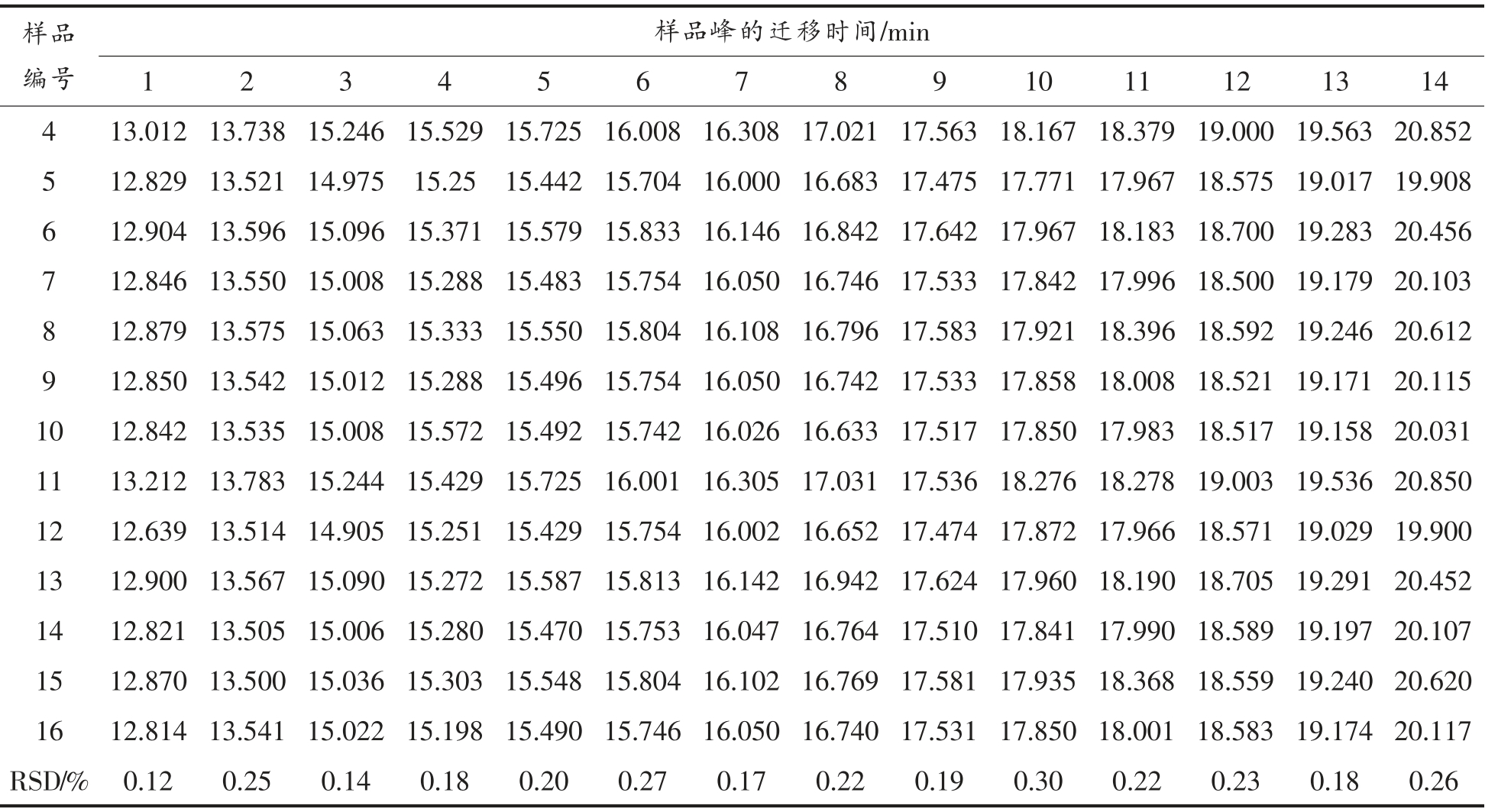
样品编号样品峰的迁移时间/min 1 2 3 4 5 6 7 8 9 10 11 12 13 14 4 13.012 13.738 15.246 15.529 15.725 16.008 16.308 17.021 17.563 18.167 18.379 19.000 19.563 20.852 5 12.829 13.521 14.975 15.25 15.442 15.704 16.000 16.683 17.475 17.771 17.967 18.575 19.017 19.908 6 12.904 13.596 15.096 15.371 15.579 15.833 16.146 16.842 17.642 17.967 18.183 18.700 19.283 20.456 7 12.846 13.550 15.008 15.288 15.483 15.754 16.050 16.746 17.533 17.842 17.996 18.500 19.179 20.103 8 12.879 13.575 15.063 15.333 15.550 15.804 16.108 16.796 17.583 17.921 18.396 18.592 19.246 20.612 9 12.850 13.542 15.012 15.288 15.496 15.754 16.050 16.742 17.533 17.858 18.008 18.521 19.171 20.115 10 12.842 13.535 15.008 15.572 15.492 15.742 16.026 16.633 17.517 17.850 17.983 18.517 19.158 20.031 11 13.212 13.783 15.244 15.429 15.725 16.001 16.305 17.031 17.536 18.276 18.278 19.003 19.536 20.850 12 12.639 13.514 14.905 15.251 15.429 15.754 16.002 16.652 17.474 17.872 17.966 18.571 19.029 19.900 13 12.900 13.567 15.090 15.272 15.587 15.813 16.142 16.942 17.624 17.960 18.190 18.705 19.291 20.452 14 12.821 13.505 15.006 15.280 15.470 15.753 16.047 16.764 17.510 17.841 17.990 18.589 19.197 20.107 15 12.870 13.500 15.036 15.303 15.548 15.804 16.102 16.769 17.581 17.935 18.368 18.559 19.240 20.620 16 12.814 13.541 15.022 15.198 15.490 15.746 16.050 16.740 17.531 17.850 18.001 18.583 19.174 20.117 RSD/% 0.12 0.25 0.14 0.18 0.20 0.27 0.17 0.22 0.19 0.30 0.22 0.23 0.18 0.26
2.3.3 电泳图谱的评价 色谱指纹图谱相似度是评价指纹图谱稳定性的有效手段之一,能够反映样品中各组分的分布比例,目前最常用的评价指标为向量夹角余弦相似度Scosθ。Scosθ 的值越接近1,样品指纹图谱与参照指纹图谱在组分分布上的一致性越高。此外,图谱间含量上的相似性常用含量相似度U 及图谱总积分峰面积与标准指纹图谱中总积分峰面积的百分比I(宏观含量相似度)两个指标评价,I 和U 越接近100%表明样品指纹图谱化学成分的含量与对照指纹图谱中化学成分的含量越相似。
式中,xti——待测图谱共有峰的峰面积;xsi——参照图谱共有峰的峰面积;n——共有峰个数;ri——样品指纹图谱中峰面积与对照图谱中对应峰面积的比值;Ai——指纹图谱中各个峰的面积;Ao——标准对照指纹图谱的峰面积总和。
被测样品的Scosθ,U 值,I 值如表3所示。由结果可知,虽然样品的个体间存在差异,但被测牛乳样品的乳蛋白电泳图谱间的相似度均大于0.900,说明被测牛乳样品的乳蛋白组成基本相似。
表3 牛乳蛋白相似度评价结果
Table 3 The similarity evaluation results of milk proteins

原料乳来源 Scosθ I/% U/% 原料乳来源 Scosθ I/% U/%1 0.965 102.3 100.2 6 0.996 85.6 104.3 2 0.954 98.0 106.0 7 0.937 101.5 93.6 3 0.990 94.3 98.9 8 0.940 96.14 115.5 4 0.948 91.8 98.0 9 0.951 99.01 106.7 5 0.939 80.9 80.0 10 0.937 88.90 97.6
2.4 原料乳蛋白掺杂检测
对分别添加不同比例的大豆分离蛋白、 水解胶原蛋白和小麦面筋蛋白的牛乳样品进行测定,将得到的电泳图与牛乳样品的电泳图进行比较,从组分的迁移时间和峰面积判断掺杂乳的情况。
2.4.1 添加大豆分离蛋白掺杂乳的检测 由图4a可知,大豆分离蛋白的出峰时间为12.0~17.0 min之间和22.542 min,由前文结果(图3f)可知,α-La、β-Lg 及α-CN(αs2-CN、αs1-CN 和αs0-CN)的出峰时间分别为13.056,13.776 min 及15.196,16.212 min 和16.888 min,大豆分离蛋白在12.0~17.0 min 之间的峰与上述3 种乳蛋白峰重合。因此,添加大豆分离蛋白后,原牛乳样品中的α-La、β-Lg 以及α-CN 溶质峰的峰面积均明显增大,通过比较发现,峰1 可以作为区别牛乳和掺入大豆分离蛋白牛乳样品的特征峰,如图4b所示。
2.4.2 添加水解胶原蛋白掺杂乳的检测 由图4d可知,水解胶原蛋白的出峰时间为11.275,15.967,16.817,17.137 min 和17.438 min,由前文结果(图3f)可知,牛乳中α-CN(αs2-CN、αs1-CN 和αs0-CN)、κ-CN、β A1-CN 及β A2-CN 的出峰时间分别为15.196,16.212 min 和16.888 min 及17.986,18.588 min 和19.202 min。如图所示,水解胶原蛋白在15.967~17.438 min 之间的峰与上述乳蛋白中的α-CN 和κ-CN 的峰重合。添加水解胶原蛋白后,通过比较,确定峰2 作为区别牛乳和添加水解胶原蛋白牛乳的特征峰(图4e)。
2.4.3 大豆分离蛋白水解胶原蛋白的掺假检出限将大豆分离蛋白和水解胶原蛋白的特征峰面积与添加比例进行线性回归分析,结果如表4所示。当大豆分离蛋白和水解胶原蛋白的添加量较大时,它们的特征峰面积与添加量之间呈正相关,线性较好。当大豆分离蛋白和水解胶原蛋白的添加量分别降至4.5%和4.8%时,其特征峰在电泳图中的面积已经很小,不能测定其峰面积。
表4 大豆分离蛋白添加量与其峰面积的关系
Table 4 The relationship between the area of the peaks and the content of soy isolate protein
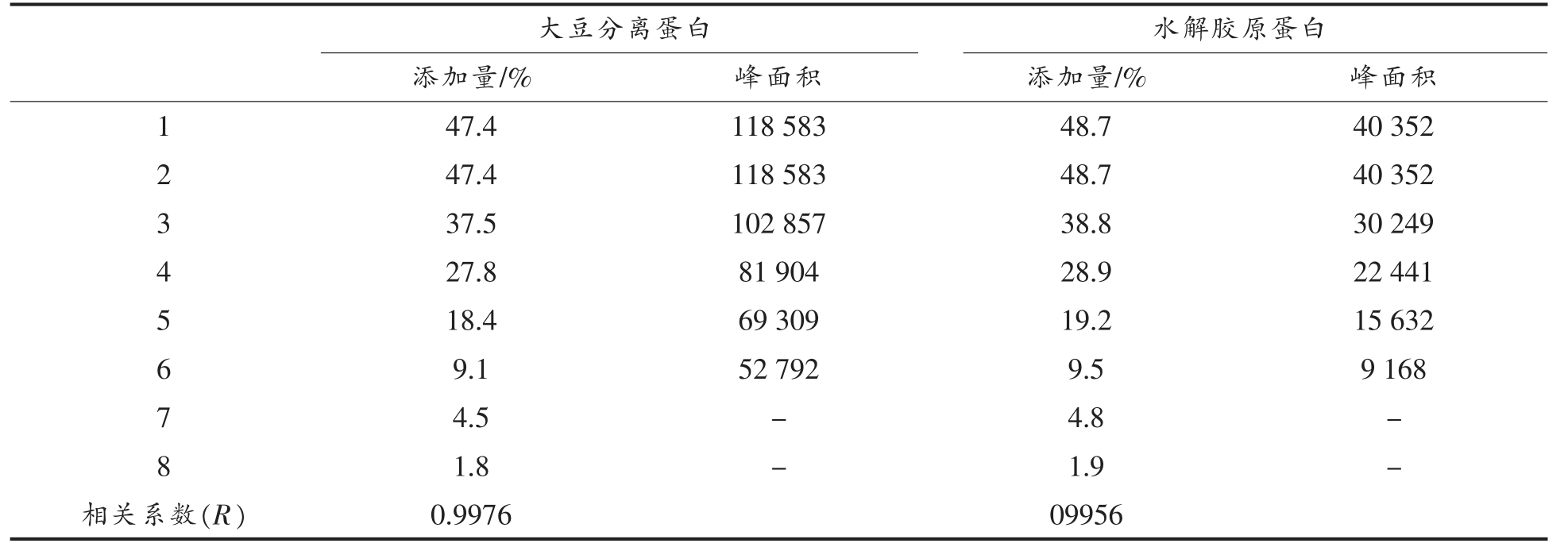
注:添加量为外源蛋白占掺杂样品总蛋白的百分比。
大豆分离蛋白 水解胶原蛋白添加量/% 峰面积 添加量/% 峰面积47.4 118 583 48.7 40 352 2 47.4 118 583 48.7 40 352 3 37.5 102 857 38.8 30 249 4 27.8 81 904 28.9 22 441 5 18.4 69 309 19.2 15 632 6 9.1 52 792 9.5 9 168 7 4.5 - 4.8 -8 1.8 - 1.9 -相关系数(R) 0.9976 09956 1
3 结论
本文通过优化毛细管区带电泳分离条件中的缓冲体系及其pH、进样条件、毛细管长度、分离电压,最终建立起适用于牛乳蛋白的最佳检测条件为选用60 cm 的毛细管柱,以pH 2.75 的磷酸盐缓冲液作为电泳缓冲液,采用6.895 kPa、10 s 的压力进样方式,分离电压设为25 kV。利用优化后的方法,以采集到的原料乳进行测定,最终建立起牛乳蛋白中14 种特征组分的毛细管区带电泳图谱。此外,建立起常见的掺假原料——大豆分离蛋白和水解胶原蛋白在本方法上的特征定性峰、 定量回归方程(相关性指数均大于0.996)以及检测限(4.5%和4.8%)。本文建立的毛细管区带电泳方法可以应用于原料乳收购环节,作为牛乳蛋白掺假的一种快速、可靠的检测方法。
参 考 文 献
[1]D′AMBROSIO C,ARENA S,SALZANO A M,et al.A proteomic characterization of water buffalo milk fractions describing PTM of major species and the identification of minor components involved in nutrient delivery and defense against pathogens[J].Protemics,2008,8(17):3657-3666.
[2]RONCADA P,GAVIRAGHI A,LIBERATORI S,et al.Identification of caseins in goat milk[J].Proteomics,2002,2(6):723-726.
[3]PISANU S,GHISAURA S,PAGNOZZI D,et al.Characterization of sheep milk fat globule proteins by two-dimensional polyacrylamide gel electrophoresis/mass spectrometry and generation of a reference map[J].Int Dairy J,2012,24(2):78-86.
[4]MIRANDA G,MAHÉ M F,LEROUX C,et al.Proteomic tools to characterize the protein fraction of Equidae milk[J].Protemics,2004,4(8):2496-2509.
[5]BONIZZI I,BUFFONI J N,FELIGINI M.Quantification of bovine casein fractions by direct chromatographic analysis of milk.Approaching the application to a real production context[J].J Chromatogr A,2009,1216(1):165-168.
[6]BONFATTI V,GRIGOLETTO L,CECCHINATO A,et al.Validation of a new reversed-phase high-performance liquid chromatography method for separation and quantification of bovine milk protein genetic variants [J].J Chromatogr A,2008,1195 (1/2):101-106.
[7]VELOSO ACA,TEIXEIRA N,FERREIRA IMPLVO.Separation and quantification of the major casein fractions by reverse -phase high -performance liquid chromatography and urea-polyacrylamide gel electrophoresis:detection of milk adulterations[J].J Chromatogr A,2002,967(2):209-218.
[8]HERRERO-MARTINEZ J M,SIMO-ALFONSO E F,RAMIS-RAMOS G,et al.Determination of cow’s milk in non-bovine and mixed cheeses by capillary electrophoresis of whey proteins in acidic isoelectric buffers[J].J Chromatogr A,2000,878(2):261-271.
[9]LUYKX D M,CORDEWENER J H,FERRANTI P,et al.Identification of plant proteins in adulterated skimmed milk powder by high-performance liquid chromatography-mass spectrometry[J].J Chromatogr A,2007,1164(1/2):189-197.
[10]CZERWENKA C,MÜLLER L,LINDNER W.Detection of the adulteration of water buffalo milk and mozzarella with cow’s milk by liquid chromatography-mass spectrometry analysis of β -lactoglobulin variants[J].Food Chem,2010,122(3):901-908.
[11]于文鹏.毛细管电泳分析蛋白质方法的初步研究[D].北京:北京林业大学,2005.
[12]SONG H,XUE H,HAN Y.Detection of cow’s milk in Shaanxi goat’s milk with an ELISA assay[J].Food Control,2011,22(6):883-887.
[13]HASELBERG R,DE JONG G J,SOMSEN G W.Capillary electrophoresis-mass spectrometry for the analysis of intact proteins[J].J Chromatogr A,2007,1159(1/2):81-109.
[14]RECIO I,AMIGO L,LÓPEZ-FANDIÑO R.Assessment of the quality of dairy products by capillary electrophoresis of milk proteins[J].J Chromatogr B,1997,697(1/2):231-242.
[15]GUTIERREZ JEN,JAKOBOVITS L.Capillary electrophoresis of α-lactalbumin in milk powders[J].J Agr Food Chem,2003,51(11):3280-3286.
[16]PATERSON G R,HILL J P,OTTER D E.Separation of β-lactoglobulin A,B and C variants of bovine whey using capillary zone electrophoresis[J].J Chromatogr A,1995,700(1/2):105-110.
[17]MIRALLES B,RAMOS M,AMIGO L.Influence of proteolysis of milk on the whey protein to total protein ratio as determined by capillary electrophoresis[J].J Dairy Sci,2003,86(9):2813-2817.
[18]IRIGOYEN A,IZCO J M,IBÁÑEZ F C,et al.Evaluation of the effect of rennet type on casein proteolysis in an ovine milk cheese by means of capillary electrophoresis[J].J Chromatogr A,2000,881(1/2):59-67.
[19]HERRERO-MARTÍNEZ J M,SIMÓ-ALFONSO E F,RAMIS-RAMOS G,et al.Determination of cow’s milk in non-bovine and mixed cheeses by capillary electrophoresis of whey proteins in acidic isoelectric buffers[J].J Chromatogr A,2000,878(2):261-271.
[20]RECIO I,AMIGO L,RAMOS M,et al.Application of capillary electrophoresis to the study of proteolysis of caseins[J].J Dairy Res,1997,64(2):221-230.
[21]HUTTERER K,DOLNÍK V.Capillary electrophoresis of proteins 2001-2003[J].Electrophoresis,2003,24(22/23):3998-4012.
[22]GARCIA -RUIZ C,TORRE M,MARINA M L.Analysis of bovine whey proteins in soybean dairylike products by capillary electrophoresis[J].J Chromatogr A,1999,859(1):77-86.
[23]RECIO I,GARCÍA-RISCO M R,AMIGO L,et al.Detection of milk mixtures in halloumi cheese[J].J Dairy Sci,2004,87(6):1595-1600.
[24]MÜLLER L,BARTÁK P,BEDNÁR P,et al.Capillary electrophoresis-mass spectrometry-a fast and reliable tool for the monitoring of milk adulteration[J].Electrophoresis,2008,29(10):2088-2093.
[25]LEE S J,CHEN M C,LIN C W.Detection of cows’ milk in goats’ milk by capillary zone electrophoresis[J].Aust J Dairy Technol,2001,56(1):24-27.
[26]MARIA PIERA CATTANEO T,NIGRO F,MARIA TOPPINO P,et al.Characterization of ewe’s milk by capillary zone electrophoresis[J].J Chromatogr A,1996,721(2):345-349.
[27]MULLER L,BARTAK P,BEDNAR P,et al.Capillary electrophoresis-mass spectrometry - a fast and reliable tool for the monitoring of milk adulteration[J].Electrophoresis,2008,29(10):2088-2093.
[28]STRICKLAND M,JOHNSON M E,BROADBENT J R.Qualitative and quantitative analysis of proteins and peptides in milk products by capillary electrophoresis[J].Electrophoresis,2001,22(8):1510-1517.
[29]RECIO I,RAMOS M,LOPEZ-FANDINO R.Capillary electrophoresis for the analysis of food proteins of animal origin[J].Electrophoresis,2001,22(8):1489-1502.
[30]HERNANDEZ -BORGES J,BORGES -MIQUEL T M,RODRIGUEZ-DELGADO M A,et al.Sample treatments prior to capillary electrophoresis -mass spectrometry[J].J Chromatogr A,2007,1153(1/2):214-226.
Optimization and Application of Capillary Zone Electrophoresis in Bovine Milk Adulteration Detection
Fang Bing1,2 Zhang Hao2 Guo Huiyuan2 Ren Fazheng2*
(1Beijing Advanced Innovation Center for Food Nutrition and Human Health,China Agricultural University,Beijing 1000832Key Laboratory of Functional Dairy,Beijing Laboratory for Food Quality and Safety,College of Food Science and Nutritional Engineering,China Agricultural University,Beijing 100083)
Abstract Objective:To establish an effective method for the rapid detection of adulteration in raw milk using the capillary electrophoresis.Methods:Optimizing the parameters of the capillary electrophoresis,including the buffer system and its pH,sampling condition,capillary length and voltage of electrophoresis.Results:The optimized condition for the rapid detection of adulteration in raw milk was:a 60 cm length capillary column,separation voltage of 25 kV,pressure sampling:6.895 kPa,10 s and a pH 2.75 phosphate buffer.14 typical milk protein peaks were determined with a good correlation index (>0.900).Furthermore,the typical peaks,qualitative equations (both with a correlation index above 0.996)and detection limits (4.5% and 4.8%,respectively)of soybean isolated protein,hydrolyzed collagen protein were established,which were two common non-bovine proteins added in milk.Conclusion:This study established an optimal capillary zone electrophoresis method in the separation and identification of raw milk,providing a fast and reliable method of bovine milk adulteration.
Keywords capillary zone electrophoresis;soybean isolated protein;collagen hydrolyzed protein;adulteration detection
文章编号 1009-7848(2019)03-0289-10
doi:10.16429/j.1009-7848.2019.03.036
收稿日期:2018-03-06
作者简介:方冰,女,1987年出生,博士,副研究员
通讯作者:任发政 E-mail:renfazheng@263.net